电催化量子化学统计力学电子结构反应动力学吉布斯自由能变d火山图计算方法涵盖、、机器学习等,工具如等助力模拟。典型内容有火山图、自由能图等,经典案例解析过电位起源。前沿聚焦动态界面、多尺度融合,推动从理论到应用的转化。
什么是电催化?
通过量子化学与统计力学方法模拟电极表面反应过程,从而预测催化剂活性、选择性及反应路径的计算科学,其核心目标是从原子尺这一领域的核心概念构建了从微观电子态到宏观反应性能的完整认知框架:dd轨道能级位置映射吸附强度:ε_d向费米能级上移时,金属与吸附物的轨道杂化增强,吸附作用变强;ε_d下移则吸附作用减弱,这一理论为通过调控活性中心电子态优化催化性能提供了明确的电子尺度依据。
d轨道的自旋状态(高自旋/低自旋)对键合强度的影响,例如在氧还原反应中,高自旋态Fe³⁺因d轨道电子排布更利于与氧中间体的π轨道形成反馈键,可有效优化、OH等中间体的吸附能,提升催化活性。
则以关键中间体的吸附能H而处于火山图顶点,这一原理为高通量筛选高活性催化剂提供了高效的理论工具。
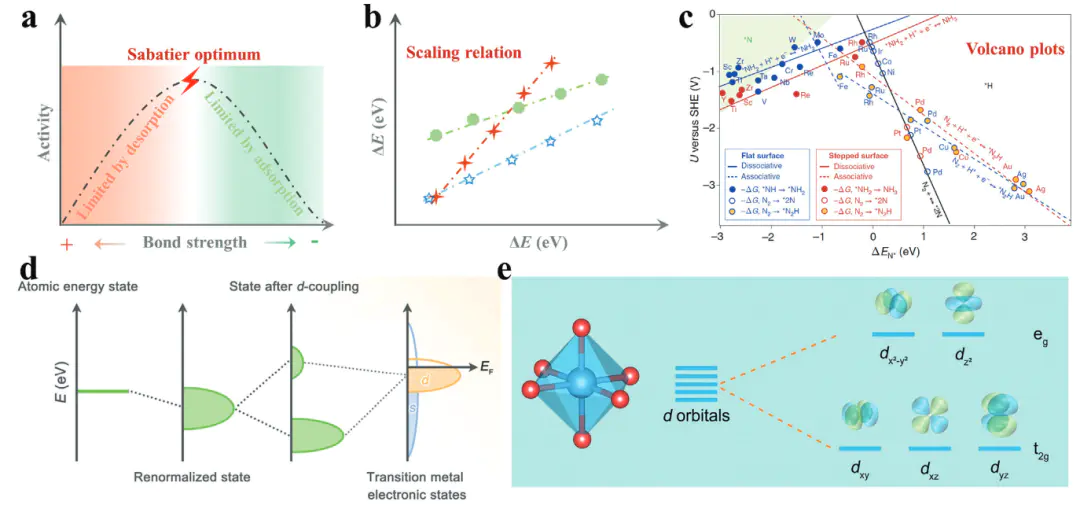
这些核心概念相互关联,共同构成了电催化理论计算的基础框架,使研究者能够从热力学、电子结构、动力学等多维度解析催化机制,推动电催化研究从经验探索向理论驱动转变。
计算方法与工具
核心计算方法中,DFT是最基础且应用最广泛的工具,其通Kohn-ShamDFT的关键模型包括计算氢电极(CHE)模型和溶剂化模型:通过将电极电位U转换为氢的化学势参考,直接建立ΔG与电极电位的关联,使理论计算可模拟不同电势下的反应行为;或则用于模拟固液界面环境,修正溶剂极性对吸附能的影响。
DOI:10.3389/fchem.2023.1286257
MD焦于动态过程模拟,通过经典力场模拟催化剂表面的动态重构、溶剂化效应及质子转移路径,AIMD则结合DFT实时计算原子间作用力,可更精准地描述电子结构与核运动的耦合,如OER中NiFeOOH表面羟基的动态演化。
包括约束通过强制反应坐标计算自由能面,元动力学通过添加偏置势加速稀有事件采样,有效识别反应路径。
ML势函数通过训练数据构建高维势能面,可实现包含10⁴以上原子的大尺度模拟,突破DFT的计算量限制,典型方法如神经网络势、高斯过程回归,在模拟纳米颗粒催化剂的尺寸效应时,计算效率比AIMD提升100倍以上。
则结合过渡态理论与动力学蒙特卡洛,通过整合计算的能垒数据,求解速率方程,模拟表面中间体覆盖度依赖的反应速率,可预测Tafel斜率和交换电流密度,例如在分析CO₂还原反应的选择性时,该方法可量化不同路径的占比,揭示选择性调控机制。
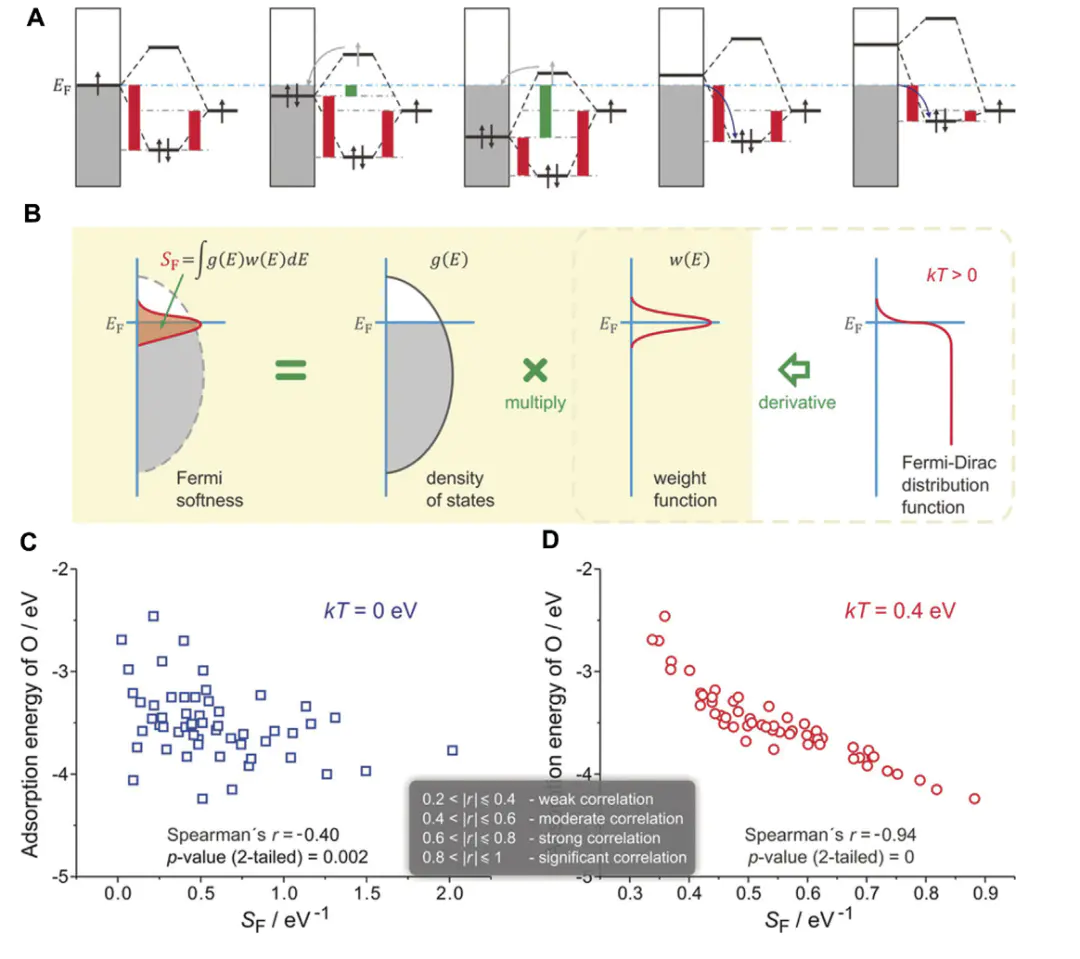
主流计算软件VASPGaussianLAMMPSaxFF等反应力场,可模拟大尺度(如微米级)固液界面的溶剂动力学,如双电层中离子的分布与迁移。
电催化理论计算的典型内容通过可视化与量化分析,直观呈现催化反应的关键特征与机制,为关联理论模型与实验现象提供了重要桥梁,火山图()源于等人的研究,其横轴为H吸附能,纵轴为交换电流密度,清晰展示了不同金属催化剂的活性分布:Pt因ΔG_H0 eV正值较大位于火山左侧,强吸附金属因ΔG_H这一结果定量验证了萨巴蒂尔原理,明确了“ΔG_HDOI自由能图()在ørskov通过计算不同电极电位下的ΔG,发现当U=1.23 V(ORR平衡电位)时,*O→*OH步骤的ΔG>0,成为速率控制步骤(RDS),对应的理论过电位η=0.45 V,与实验测量值高度吻合。
OHOHDOI动态表面重构模拟在等的研究中展现了其价值,通过AIMD模拟OER过程中NiFeOOH表面的羟基演化,发现Fe位点的引入会诱导周围O原子的电子云重分布,促进O-O键耦合:模拟显示,Fe³⁺的高自旋态使相邻O原子的电子密度降低,更易与另一*O形成新键,将O-O耦合能垒从纯NiOOH的0.8 eV降至0.5 eV。
DOI:10.1002/smsc.202100011
ORR理论过电位起源
等的论文“Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode”,ORRDOI:10.1021/jp047349j
DFT-PW91OH,并计算其吸附能,为后续自由能分析奠定基础。
计算引入电极电位的影响,将电子结构计算与电化学电位关联,绘制了U=0 V和U=1.23 V时的自由能图。
O→OH动力学描述符拓展部分,建立了吸附能与ORR活性的火山关系,发现过渡金属的催化活性随ΔG_OH变化呈现火山型分布,Pt因ΔG_OH处于最优范围而位于火山顶点,这一关系不仅解释了不同金属的活性差异,更提供了通过调控ΔG_OH筛选高活性ORR催化剂的通用方法。
首次将电极电位引入自由能计算,构建了“电位依赖自由能”的分析框架,使理论计算能够直接关联电化学实验中的可测量,HER同时,该研究确立了“中间体吸附能作为活性描述符”的研究范式,通过关键中间体的吸附能预测催化活性,大幅简化了催化剂筛选的复杂度,为后续高通量计算与机器学习辅助设计奠定了基础。
DOI:10.1021/jp047349j
电催化前沿
动态界面建模是当前的核心挑战,传HER电场效应通过改变表面电荷分布调制吸附强度,而表面重构会实时改变活性位点的配位环境,这些动态因素的耦合模拟需要更高效的AIMD算法与更大的计算尺度,目前通过增强采样技术可部分捕捉关键动态事件,但全周期模拟仍受限于计算成本。
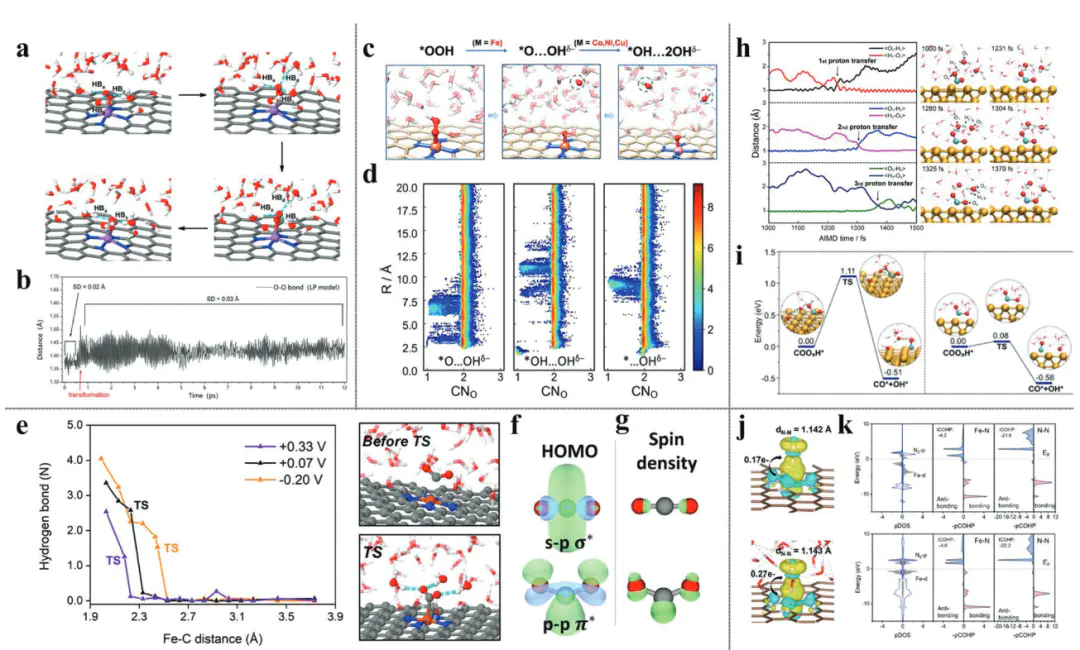
多尺度方法融合是提升计算效率与范围的关键路径,机器学习(构建的ML势,可在保持DFT精度的前提下,将模拟尺度扩展至10⁴原子以上,模拟时间从皮秒级延长至纳秒级,例如在模拟纳米多孔催化剂的传质效应时,ML势加速的MD可捕捉离子在孔道内的扩散路径,而这是传统DFT无法实现的。
DOI:10.3389/fchem.2023.1286257
–开发非绝热动力学方法可ORR此外,自旋轨道耦合(SOC)效应在重元素催化剂(如Ir、Pt)中的影响逐渐受到关注,SOC可分裂d轨道能级,改变中间体的吸附构型,例如在IrO₂的OER中,SOC使*O的吸附能降低0.08 eV,影响过电位计算。
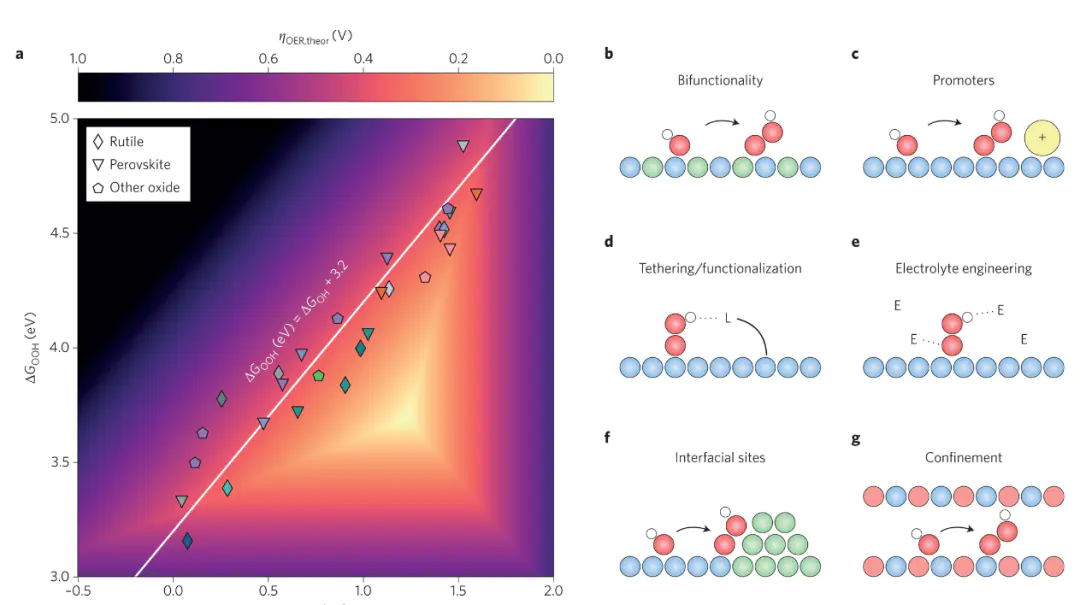
这些前沿方向的发展,正推动电催化理论计算从“静态、单尺度、热力学主导”向“动态、多尺度、动力学精准”转变,通过“描述符指导设计”的逆向范式,加速高活性、高选择性电催化剂的开发,推动电催化在燃料电池、电解水、CO₂转化等领域的应用突破。
总结
电催化理论计算以量子力学为基础,通过密度泛函理论、分子动力学、机器学习等多方法的协同联动,构建了从电子结构解析到宏观催化性能预测的跨尺度理论体系,为“催化剂基因工程”——即通过原子级调控实现催化性能精准设计——提供了核心支撑这一体系的核心价值在于打破了电催化研究的经验主义局限:从电子层面揭示活性中心的d带中心、自旋态等电子结构参数与中间体吸附能的关联,建立了“结构–电子态–吸附能”的映射关系。
通过典型计算内容,将抽象的与可观当前,电催化理论计算正朝着动态界面模拟、多尺度融合、动力学精准化的方向发展,其在非贵金属催化剂开发、单原子催化剂设计、新型反应路径探索等领域的应用持续取得突破。
随着计算方法的不断完善与算力的提升,电催化理论计算将实现从“解释现象”到“预测性能”再到“设计材料”的全链条跨越